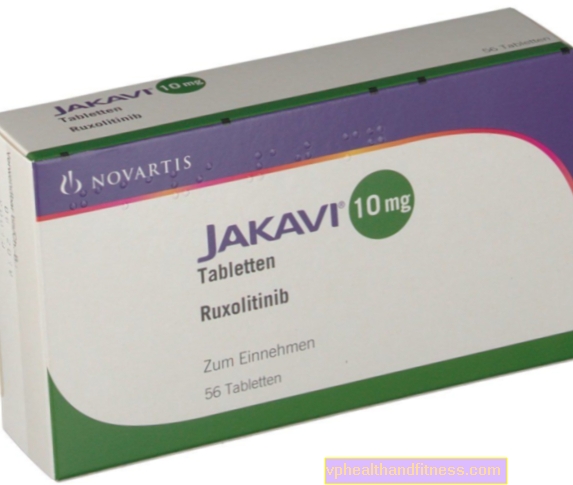
1 tablett innehåller 5 mg, 10 mg, 15 mg eller 20 mg ruxolitinib som fosfat. Beredningen innehåller laktos.
| namn | Förpackningens innehåll | Den aktiva substansen | Pris 100% | Senast ändrad |
| Jakavi | 56 st, bord | Ruxolitinib | 2019-04-05 |
Handling
Ett läkemedel mot cancer, en proteinkinashämmare. Ruxolitinib är en selektiv hämmare av Janus kinaser (JAK), JAK1 och JAK2, som förmedlar signalering för ett antal cytokiner och tillväxtfaktorer som spelar en viktig roll vid hemopoies och immunfunktion. Det kännetecknas av hög permeabilitet, god löslighet och snabb frisättning. Det absorberas snabbt efter oral administrering, Cmax uppnås cirka 1 timme efter dosering. Bindningen till plasmaproteiner är cirka 97%, huvudsakligen till albumin. Ruxolitinib passerar inte blod-hjärnbarriären. Det metaboliseras huvudsakligen av CYP3A4, med ytterligare bidrag från CYP2C9. I plasma är läkemedlet huvudsakligen närvarande som oförändrat läkemedel och som två aktiva metaboliter. Ruxolitinib elimineras huvudsakligen genom metabolism. Den genomsnittliga eliminationen av T0.5 för ruxolitinib är cirka 3 timmar, det utsöndras huvudsakligen i urin och avföring.
Dosering
Oralt. Behandling bör endast initieras av en läkare med erfarenhet av administrering av läkemedel mot cancer. Ett fullständigt blodtal med ett test av vita blodkroppar bör utföras innan behandlingen påbörjas. En fullständig blodräkning med en vit blodkroppsutstrykning bör utföras var 2-4 veckor tills dosen är stabiliserad, och sedan beroende på kliniska indikationer. Initial dos. Myelofibros: 15 mg två gånger dagligen hos patienter med trombocytantal mellan 100 000 / mm3 och 200 000 / mm3 och 20 mg två gånger dagligen hos patienter med trombocytantal> 200 000 / mm3. Polycytemia Vera: 10 mg oralt 2 gånger om dagen. Det finns begränsad information om den rekommenderade startdosen för patienter med trombocytantal mellan 50 000 / mm3 och 3 - max. startdosen som rekommenderas för dessa patienter är 5 mg två gånger dagligen och bör ökas med försiktighet. Dosändringar. Doser kan justeras utifrån läkemedlets säkerhet och effektivitet. Behandlingen bör avbrytas om trombocytantalet är mindre än 50 000 / mm3 eller det absoluta antalet neutrofiler är mindre än 500 / mm3. Behandlingen bör också avbrytas hos PV-patienter när hemoglobinnivåerna är under 8 g / dL. Efter att ha ökat antalet blodvärden över dessa värden kan doseringen återupptas med en dos på 5 mg två gånger dagligen med gradvis ökning baserat på resultaten av ett fullständigt blodprov med utstryk. Dosreduktion bör övervägas om trombocytantalet faller under 100 000 / mm3 för att undvika avbrytande av behandlingen på grund av trombocytopeni. Dosreduktion bör också övervägas hos PV-patienter om hemoglobin är under 12 g / dL, och dosreduktion rekommenderas om hemoglobin är under 10 g / dL. Om behandlingen anses vara otillräckligt effektiv och blodantalet är tillräckligt, kan dosen ökas med maximalt 5 mg två gånger dagligen, upp till max. doser på 25 mg två gånger dagligen. Startdosen bör inte ökas under de första fyra veckorna av behandlingen och ska därefter inte ökas oftare än med två veckors intervall. Max. beredningsdosen är 25 mg två gånger om dagen. Dosjusteringar när du tar samtidigt starka CYP3A4-hämmare eller flukonazol. En enhetsdos bör minskas med cirka 50% och administreras två gånger om dagen. Samtidig användning av preparatet med flukonazol i doser över 200 mg dagligen bör undvikas. Under behandling med starka hämmare av CYP3A4 eller dubbla hämmare av CYP2C9- och CYP3A4-enzymer rekommenderas mer frekvent (t.ex. två gånger i veckan) hematologiska parametrar och tecken och symtom på läkemedelsrelaterade biverkningar. Avbrytande av behandlingen. Behandlingen bör fortsätta så länge nytta-riskbalansen förblir positiv, men bör avbrytas efter 6 månader om det inte har skett någon minskning av mjältstorleken eller symtomförbättring sedan behandlingen startade. Det rekommenderas att patienter som uppvisar en viss grad av klinisk förbättring ska avbryta behandlingen med ruxolitinib om de upplever 40% mjälteförlängning jämfört med baslinjelängden (ungefär motsvarande 25% mjältexpansion) och ingen verklig förbättring av mjälten observeras. i förhållande till symtomen associerade med sjukdomen. Särskilda grupper av patienter. Ingen speciell dosjustering krävs för patienter med lätt eller måttligt nedsatt njurfunktion. Hos patienter med svårt nedsatt njurfunktion (CCr 3 till 200 000 / mm3. En enstaka dos på 20 mg eller 2 doser på 10 mg med 12 timmars mellanrum rekommenderas för MF-patienter med trombocytantal> 200 000 / mm3. Efterföljande doser (enstaka administrering eller 2 doser på 10 mg med 12 timmars mellanrum) bör endast ges på hemodialysdagar efter varje dialysperiod. Rekommenderad startdos för hemodialyspatienter med ESRD och PV är en enstaka dos på 10 mg eller två doser efter varje dialysperiod. 5 mg ges med 12 timmars intervall efter dialys och endast på hemodialysdag. Dessa doseringsrekommendationer är simulerade och all dosjustering hos ESRD-patienter ska noggrant övervakas för säkerhet och effekt. Inga data finns tillgängliga för patienter som genomgår behandling. peritonealdialys eller kontinuerlig venovänös hemofiltrering Hos patienter med nedsatt leverfunktion är den rekommenderade startdosen baserad på och trombocyter bör minskas med cirka 50% och ges två gånger dagligen. Efterföljande doser bör justeras baserat på läkemedlets säkerhet och effektivitet. Patienter som diagnostiserats med leversvikt under behandlingen med preparatet ska ha ett fullständigt blodprov med utstryk minst 1 på 1-2 veckor under de första 6 veckorna efter påbörjad behandling och sedan efter stabilisering av leverfunktion och blodprov - om tillgängligt kliniska indikationer. Dosen kan justeras för att minska risken för cytopeni. Ytterligare dosjusteringar hos äldre rekommenderas inte. Säkerhet och effekt hos barn under 18 år har inte fastställts. Det finns inga tillgängliga data. Sätt att ge. Ta med eller utan mat. Om en dos saknas ska patienten inte ta en extra dos utan ta nästa föreskrivna dos.
Indikationer
Marv fibros. Behandling av splenomegali associerad med en sjukdom eller symtom som ses hos vuxna patienter med primär benmärgsfibros (även känd som kronisk idiopatisk benmärgsfibros), myelofibros som föregås av polycytemi (hyperemi) vera eller benmärgsfibros som föregås av essentiell trombocytemi. Tuftad tuftad anka. Behandling av vuxna patienter med polycytemi vera som är resistenta eller intoleranta mot hydroxikarbamidbehandling.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne. Graviditet och amning.
Försiktighetsåtgärder
Läkemedlet kan orsaka hematologiska biverkningar inklusive trombocytopeni, anemi och neutropeni, därför krävs ett fullständigt blodprov med vita blodkroppar innan du påbörjar behandlingen. Behandlingen ska avbrytas hos patienter med trombocytantal mindre än 50 000 / mm3 eller absolut neutrofilantal mindre än 500 / mm3. Det har noterats att patienter med lågt antal blodplättar (3) vid tidpunkten för behandlingen inleds är mer benägna att utveckla trombocytopeni under behandlingen. Trombocytopeni är i allmänhet reversibel och kan vanligtvis hanteras genom att minska dosen eller tillfälligt stoppa beredningen, även om trombocyttransfusioner kan vara nödvändiga beroende på den kliniska indikationen. Patienter som utvecklar anemi kan behöva blodtransfusioner, och dosjustering eller avbrytande av behandlingen kan övervägas hos patienter med anemi. Patienter med en hemoglobinnivå under 10,0 g / dL i början av behandlingen löper större risk för hemoglobinnivåer under 8,0 g / dL under behandlingen jämfört med patienter med högre hemoglobinnivåer vid baslinjen, därför hos patienter med hemoglobinnivåer vid baslinjen. hemoglobin under 10,0 g / dl rekommenderas frekventare övervakning av hematologiska parametrar samt bedömning av tecken och symtom som indikerar biverkningar i samband med användning av preparatet. Neutropeni (absolut neutrofilantal <500 / mm3) var i allmänhet reversibel och hanterbar genom att tillfälligt hålla läkemedlet. Ett fullständigt blodprov bör utföras så ofta som kliniskt indikerat och dosjustering efter behov. Allvarliga bakterie-, mykobakterie-, svamp-, virus- och andra opportunistiska infektioner har inträffat hos patienter som behandlats med preparatet, därför ska patienter bedömas med avseende på risken för allvarliga infektioner. Läkare bör noga övervaka patienter som får läkemedlet med avseende på tecken och symtom på infektioner och inleda lämplig behandling omedelbart. Behandlingen bör inte startas förrän det inte längre finns någon allvarlig aktiv infektion. Tuberkulos har rapporterats hos patienter som tar medicinen mot myelofibros och patienter bör testas för aktiv eller inaktiv (latent) tuberkulos innan behandlingen påbörjas, enligt lokala rekommendationer. Undersökningar bör omfatta sjukdomshistoria, möjliga tidigare kontakter med tuberkulospatienter och / eller lämpliga screeningtester, såsom lungröntgen, tuberkulintest och / eller, om tillämpligt, interferon-γ-frisättningstest. Förskrivare bör komma ihåg risken för ett falskt negativt tuberkulinhudtest, särskilt hos svårt sjuka eller immunförsvarade patienter. Ökningar av hepatit B-virus (HBV-DNA-titer), med eller utan samtidig ökning av ALAT och AST, har rapporterats hos patienter med kronisk HBV-infektion som tar detta läkemedel. Effekten av läkemedlet på viral replikation hos patienter med kronisk HBV-infektion är okänd; kroniska HBV-patienter ska behandlas och övervakas enligt kliniska riktlinjer. På grund av risken för bältros bör läkare instruera patienter att känna igen de tidiga tecknen och symtomen och rekommendera att behandlingen påbörjas så tidigt som möjligt. Progressiv multifokal leukoencefalopati (PML) har rapporterats vid användning av Jakavi för behandling av MF, därför bör läkare vara vaksamma när det gäller symtom som kan tyda på PML som patienter kanske inte märker (t.ex. kognitiv, neurologisk eller mental). Patienter bör övervakas för nya eller förvärrade symtom och om sådana symtom utvecklas bör man överväga att hänvisa patienten till en neurolog eller vidta lämpliga diagnostiska motåtgärder. Om PML misstänks ska ytterligare behandling avbrytas tills PML har uteslutits. Icke-melanom maligna hudsvulster (NMSC) (inklusive basalcellscancer, skivepitelcancer och Merkelcellkarcinom) har rapporterats hos patienter som behandlats med ruxolitinib, de flesta av dessa patienter har haft långvarig hydroxikarbamidbehandling och tidigare NMSC eller tidigare cancerläkemedel. Periodisk hudundersökning rekommenderas hos patienter med ökad risk för hudcancer. Behandling med beredningen var associerad med ökningar av lipidparametrar, inklusive totalt kolesterol, HDL-kolesterol, LDL-kolesterol och triglycerider - det rekommenderas att övervaka lipidnivåer och behandla dyslipidemi enligt kliniska riktlinjer. Startdosen bör minskas hos patienter med svårt nedsatt njurfunktion. Hos patienter med njursjukdom i slutstadiet och MF som får hemodialys bör startdosen baseras på trombocytantal. efterföljande doser bör endast administreras hemodialysdagen efter slutet av varje hemodialysperiod. Ytterligare justeringar av dosen bör göras med noggrann övervakning av läkemedlets säkerhet och effektivitet. Startdosen bör minskas med cirka 50% hos patienter med nedsatt njurfunktion. Ytterligare dosjusteringar bör göras på grundval av läkemedelssäkerhet och effekt. Om preparatet ska administreras samtidigt med starka hämmare av CYP3A4 eller dubbla hämmare av CYP3A4- och CYP2C9-enzymer (t.ex. flukonazol) ska enhetsdosen minskas med cirka 50% och administreras två gånger dagligen. Samtidig användning av cytoreduktiva eller hematopoetiska tillväxtfaktordroger och preparatet har inte studerats. Efter avbrott eller avbrytande av behandlingen kan MF-symtom uppträda igen inom cirka en vecka. Det finns kända fall av patienter som avbrutit behandlingen med preparatet upplever allvarligare händelser, särskilt de med annan akut comorbid sjukdom. Det är inte känt om abrupt avbrytande av behandlingen har bidragit till förekomsten av dessa händelser. Gradvis avsmalnande av dosen kan övervägas förutom när abrupt avbrytande av behandlingen är nödvändig, även om användningen av att avta dosen inte har bevisats. Beredningen innehåller laktos - ska inte användas till patienter med sällsynta ärftliga problem med galaktosintolerans, Lapp-laktasbrist eller malabsorption av glukos-galaktos.
Oönskad aktivitet
Hos patienter med polycytemi vera. Mycket vanliga: urinvägsinfektioner, CTCAE grad 3 (3) och grad 3 (50 000 - 25 000 / mm3) anemi, grad 3 neutropeni.(3) och 4 (3) CTCAE, intrakraniell blödning, gastrointestinal blödning, annan blödning (inklusive epistaxis, postoperativ blödning och hematuri), gas, CTCAE grad 3 alaninaminotransferas förhöjning ( > 5x - 20x ULN). Mindre vanliga: tuberkulos. Hos patienter med myelofibros. Mycket vanliga: CTCAE-anemi av vilken typ som helst, trombocytopeni av CTCAE-klass av vilken grad som helst, blödning (blödning inklusive intrakraniell och gastrointestinal blödning, blåmärken och annan blödning, blåmärken, annan blödning (inklusive epistax, post-procedurell och hematuri), CTCAE grad 1 och 2 hyperkolesterolemi, CTCAE grad 1 hypertriglyceridemi, yrsel, CTCAE grad 1 alanin och aspartat förhöjning av aminotransferas, högt blodtryck Vanliga: urinvägsinfektioner, herpes zoster, grad 3 trombocytopeni (50 CTCAE 000-25000 / mm3), viktökning, förstoppning Mindre vanliga: CTCAE grad 3 (3) anemi förhöjt alaninaminotransferas (> 5x - 20x ULN). Med MF kan återfall av MF-symtom som trötthet, benvärk, feber, klåda, nattliga svettningar, symtomatisk förstoring av mjälten och viktminskning inträffa. I kliniska prövningar med MF återgick den totala poängen för MF-symptom gradvis till baslinjen inom 7 dagar efter avslutad behandling.
Graviditet och amning
Användningen av preparatet under graviditeten är kontraindicerat. Fertila kvinnor bör använda effektiva preventivmetoder under behandlingen, och om en kvinna blir gravid under behandlingen bör en individuell risk-nytta-bedömning göras med rådgivning om den möjliga risken för fostret. Det får inte användas under amning och därför bör amning avbrytas när behandlingen påbörjas.
Kommentarer
Har ingen eller försumbar lugnande effekt. Patienter som upplever yrsel efter att ha tagit preparatet bör dock avstå från att köra bil eller använda maskiner.
Interaktioner
Interaktionsstudier har endast utförts på vuxna. Ruxolitinib elimineras genom metabolism katalyserad av CYP3A4 och CYP2C9, därför kan preparat som hämmar aktiviteten hos dessa enzymer resultera i ökad exponering för ruxolitinib. Vid administrering av läkemedlet med starka hämmare av CYP3A4 (såsom, men inte begränsat till, iboceprevir, klaritromycin, indinavir, itrakonazol, ketokonazol, lopinavir / ritonavir, ritonavir, mibefradil, nefazodon, nelfinavir, posaconazol, saquinavir, en enhet telavilap) 50% och administreras två gånger om dagen. Patienter bör övervakas noggrant (t.ex. två gånger i veckan) för eventuell cytopeni, och dosen bör gradvis ökas baserat på säkerhet och effekt. En 50% dosreduktion bör övervägas vid användning av preparat som är dubbla hämmare av CYP2C9- och CYP3A4-enzymer (t.ex. flukonazol). Samtidig användning av läkemedlet med flukonazol i doser över 200 mg per dag bör undvikas. När man tar inducerare av CYP3A4 (såsom men inte begränsat till avasimib, karbamazepin, fenobarbital, fenytoin, rifabutin, rifampin (rifampicin), johannesört (Hypericum perforatum), bör patienter övervakas noggrant och dosen bör gradvis ökas baserat på säkerhet och effekt. Ingen dosjustering rekommenderas när ruxolitinib administreras samtidigt med milda till måttliga hämmare av CYP3A4 (såsom, men inte begränsat till, ciprofloxacin, erytromycin, amprenavir, atazanavir, diltiazem, cimetidin), men patienter bör övervakas noggrant för eventuell cytopeni när behandling med måttliga hämmare påbörjas. CYP3A4: Ruxolitinib kan hämma P-glykoprotein och bröstcancerresistensprotein (BCRP) i tarmen, vilket kan öka den systemiska exponeringen av substrat för dessa transportörer, såsom dabigatranetexilat, cyklosporin, rosuvastatin och potentiellt digoxin. läkemedelsövervakning (TDM) eller kliniskt tillstånd efter administrering av nämnda ämnen. Det är möjligt att den potentiella hämningen av P-gp och BCRP i tarmen kommer att minimeras om tiden mellan läkemedelsadministrationer hålls så lång som möjligt. Samtidig användning av hematopoietiska tillväxtfaktorer och preparatet har inte studerats. Det är inte känt om hämning av Janus-kinaser (JAK) av Jakavi minskar effektiviteten hos hematopoietiska tillväxtfaktorer eller om hematopoietiska tillväxtfaktorer påverkar preparatets effektivitet. Samtidig användning av cytoreduktiv behandling och preparatet har inte studerats - säkerheten och effekten av samtidig administrering av dessa läkemedel är okänd. Ruxolitinib hämmar inte metabolismen av det orala CYP3A4-substratet midazolam - därför förväntas ingen ökning av exponeringen för CYP3A4-substrat när dessa läkemedel administreras tillsammans med Jakavi. Beredningen påverkar inte farmakokinetiken för ett p-piller innehållande etinylöstradiol och levonorgestrel, därför förväntas effekten av preventivmedel som innehåller denna kombination inte minskas vid samtidig användning av ruxolitinib.
Beredningen innehåller ämnet: Ruxolitinib
Ersatt läkemedel: NEJ



















---pseudochoroba-czy-rzeczywiste-zagroenie-wywiad.jpg)