Spongiform encefalopati (prionsjukdomar) är de sjukdomar där patologiska former av prionproteiner är involverade i utvecklingen. Vi vet mer och mer om prionsjukdomar, men de viktigaste aspekterna förblir okända - för närvarande har medicin inte medel för att bota patienter från dessa sjukdomar.
Spongiforma encefalopatier, dvs. prionsjukdomar, kan utvecklas under livet, medan andra härrör från ärftliga genmutationer från födseln. Inom denna grupp finns det flera enheter som förekommer hos människor, exempelvis Creutzfeldt-Jakobs sjukdom eller dödlig familjär sömnlöshet.
Prionsjukdomar har länge varit mycket mystiska. Till skillnad från andra patogener, som bakterier, virus eller svampar, innehåller de inte nukleinsyra - prioner är endast gjorda av proteiner. Teorin om prionsjukdomar upptäcktes av S. Prusiner, denna upptäckt uppskattades mycket i det vetenskapliga samfundet - 1997 fick forskaren Nobelpriset i medicin. Även om relativt många år har gått sedan prion-konceptet föddes, tror vissa forskare fortfarande att det är ofullständigt och undersöker vidare dessa tillstånds natur - men några av de faktorer som är ansvariga för spongiform encefalopati har redan bekräftats.
Prionsjukdomar: orsaker
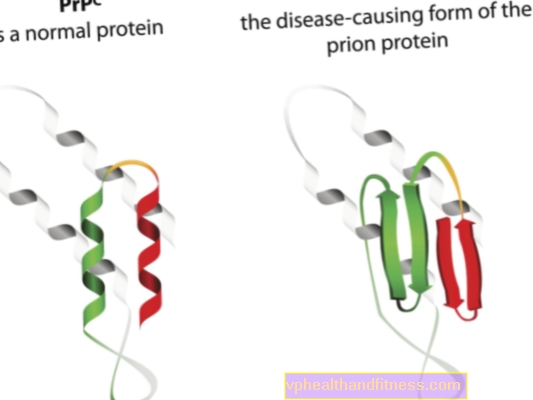
Etiologin hos prionsjukdomar är relaterad till omvandlingen av normala prionproteiner till patogena, patogena former. Prioner är proteinmolekyler som finns i varje människas kropp. Deras funktion är inte helt klar ännu, men det är känt att prionproteiner under normala förhållanden inte skadar kroppen. Situationen är annorlunda när prioner förändrar sin struktur och blir patogena partiklar - då utvecklas en av flera spongiforma encefalopatier. Prioner som naturligt förekommer i kroppen kallas PRPC, medan onormala former kallas PRPSC. De senare är ett allvarligt problem inte bara för att de kan ackumuleras i nervvävnaden i form av avlagringar och generera dess skador, utan också för att de har förmågan att förvandla normala prioner till en missformad form (helt enkelt uttryckt, PRPSC kan "infektera" normala proteiner med sin patogena potential).
Läs också: Huntingtons sjukdom (Huntingtons chorea): orsaker, symtom, behandling Muskelskakningar - orsaker. Vad betyder muskel tremor? Sjukdomar som dödar snabbast: CHOCK, EBOLA, DAMN, ATTACK, EMERGENCY [GALE ...I grund och botten finns det 3 orsaker till spongiform encefalopati:
- sporadisk (patogen mutation förekommer i somatiska celler, den inträffar under patientens liv),
- familj (som härrör från mutationer som ärvts från föräldrar),
- Passage (relaterad till införandet av patogena prioner i människokroppen, t.ex. genom tillväxthormonpreparat kontaminerade med dessa partiklar eller hornhinnetransplantation från en person som lider av någon svampformad encefalopati).
Spongiform encefalopati: Creutzfeldt-Jakobs sjukdom
Creutzfeldt-Jakobs sjukdom (CJD) beskrevs först i början av 1920-talet. Det finns fyra typer av sjukdomen:
- sporadisk CJD (den vanligaste, står för upp till 9/10 av alla CJD-fall)
- hemstad CJD
- överväldigad av CJD
- variant av CJD
Den kliniska bilden under olika varianter av Creutzfeldt-Jakobs sjukdom kan variera. De vanligaste sjukdomarna under denna grupp av svampformade encefalopatier är:
- demenssjukdomar (inklusive progressiv försämring av minne, uppmärksamhet och koncentration)
- myoklonus (ofrivilliga rörelser som plötsliga ryck i musklerna)
- cerebellär dysfunktion (manifesterad till exempel av balansstörningar)
- suddig syn
- pyramidala och extrapyramidala symtom
Under CJD-varianter kan psykiska störningar (t.ex. ångest, deprimerat humör), smärta och andra ofrivilliga rörelser än de som nämns ovan också förekomma.
Prognosen för Creutzfeldt-Jakobs sjukdom är dålig - till exempel hos patienter med sporadisk CJD tar det i genomsnitt fyra till fem månader från sjukdomssymtom till döds.
Spongiform encefalopati: Gerstmann-Straussler-Scheinkers syndrom
Gerstmann-Straussler-Scheinkers syndrom (GSS) löper vanligtvis i familjer och orsakas av en ärftlig mutation i PRNP-genen. Det anses vara den långsammaste utvecklingen av spongiform encefalopati. GSS-teamet inkluderar:
- spinocerebellär ataxi
- dysartri
- demenssjukdomar
- sväljningsstörningar
- nystagmus
- ökad muskelspänning
Patienter som diagnostiserats med GSS har varierande tid, och hos vissa patienter inträffar död mer än 10 år efter starten.
Spongiform encefalopati: dödlig familjär sömnlöshet
Dödlig familjär sömnlöshet är en prionsjukdom orsakad av mutationer i PRNP-genen. Sjukdomen är extremt sällsynt och har hittills diagnostiserats i 28 familjer över hela världen. Under dödlig familjär sömnlöshet är det första symptomet oförmåga att sova. Detta problem resulterar i ångeststörningar och att patienten upplever hallucinationer. Effekten av den ständiga bristen på nattvila är dysfunktion i det autonoma systemet (inklusive förändringar i hjärtfunktion, svettningar och matsmältningssjukdomar), det finns också en progressiv minskning av kroppsvikt. I mer avancerade stadier av dödlig familjär sömnlöshet uppträder hormonella störningar och symtom på demens uppträder under sjukdomsförloppet.
Prognosen för dödlig familjär sömnlöshet, som för andra spongiforma encefalopatier, är dålig: patienter dör vanligtvis inom tre år efter uppkomsten.
Spongiform encefalopati: prionopati med varierande känslighet för proteas
Förekomsten av de diskuterade spongiforma encefalopatierna är främst relaterad till mutationer i PRNP-genen. Dessa mutationer gäller emellertid olika kodoner av denna gen, och därför skiljer sig flera olika prionsjukdomar. En relativt nyligen beskriven (2008) enhet är prionopati med varierande känslighet för proteas. Människor som lider av denna sjukdom bär mutationer i så många som tre kodoner av PRNP-genen.
Vid prionopati med varierande känslighet för proteas upplever patienter:
- kognitiv försämring
- extrem svårighetsgrad av psykiatriska störningar: de kan vara eufori och agitation, men också betydande apati
- dysartri
- afasi (störningar i språkfunktioner)
Den genomsnittliga varaktigheten av sjukdomen i denna prionopati är mindre än 4 år.
Spongiform encefalopati: kuru
Kuru anses nu vara en sjukdom som praktiskt taget inte existerar längre - den hittades i representanter för stammar från Papua Nya Guinea, som utövade kannibalistiskt beteende. Det dominerande symptomet på denna spongiform encefalopati är progressiv cerebellär ataxi. Det kan åtföljas av ofrivilliga rörelser (främst i form av korea, skakningar och atetos), samt urin- och fekal inkontinens. Patienter på kuru upplever också betydande humörsvängningar, de utvecklar primitiva reflexer (t.ex. sugande). Ett ganska karakteristiskt problem i fallet med denna prionsjukdom är tvingade gråt eller skratt - på grund av det senare fenomenet kallas kuru ibland för "skrattdöd".
Spongiform encefalopati: diagnos
Prionsjukdomar kan misstänkas på grundval av patientens symptom. De är dock ganska ospecifika, eftersom de också kan förekomma under ett antal andra sjukdomar som inte är relaterade till prioner. Av denna anledning används även följande vid diagnos av spongiforma encefalopatier:
- avbildningstest (t.ex. magnetisk resonansavbildning, som gör det möjligt att upptäcka förändringar relaterade till degenerering av hjärnan av prionproteiner)
- laboratorietester (såsom bedömning av proteinkoncentrationer i cerebrospinalvätskan, t.ex. MAP-tau, S-100 eller 14-3-3 proteiner),
- genetiska tester (för att upptäcka närvaron av mutationer hos patienten),
- immunhistokemiska tester (med antikroppar mot prionproteiner).
Diagnosen kan också bekräftas genom obduktion av hjärnan, där det är möjligt att hitta förändringar som är karakteristiska för svampformade encefalopatier. Dessa kan vara svampiga skador, fördelade på olika sätt och med en annan struktur (beroende på en specifik sjukdomsenhet) amyloida plack och neuronal defekter.
Spongiform encefalopati: behandling
Prionsjukdomar är för närvarande obotliga - trots många studier som har pågått i många år har medicinen fortfarande inga läkemedel som kan sakta ner eller helt hämma deras framsteg. Symptomatisk behandling används hos patienter med spongiform encefalopati, som syftar till att lindra symtomens intensitet och förbättra deras livskvalitet så mycket som möjligt.
Arbetet med att behandla spongiforma encefalopatier pågår dock fortfarande. Forskare försöker använda olika metoder - det första exemplet är genterapi. De skulle påverka nukleinsyror och mutationerna som finns i deras struktur - syftet med genterapi skulle vara att neutralisera fel i den genetiska koden. Ett annat tillvägagångssätt är grunden för immunterapi - arbete pågår för att skapa antikroppar vars roll skulle vara att eliminera patogena prioner. En annan metod som ser potentialen att bekämpa spongiforma encefalopatier är behandling med användning av syntetiserade proteinmolekyler, som en gång introducerats i patientens kropp skulle neutralisera patologiska proteiner.
Rekommenderad artikel:
Encefalopatier - orsaker, typer och symtom








---ywienie-i-opieka-ile-yje-winka-morska.jpg)