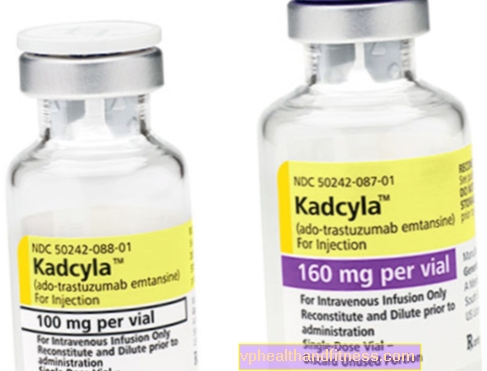
1 injektionsflaska innehåller 100 mg (160 mg) pulver för beredning 5 ml (8 ml) trastuzumab emtansinkoncentratlösning i en koncentration av 20 mg / ml för infusion efter beredning.
| namn | Förpackningens innehåll | Den aktiva substansen | Pris 100% | Senast ändrad |
| Kadcyla | 1 injektionsflaska, pulver för beredning Slutgiltig lösning till inf. | Trastuzumab emtansin | 2019-04-05 |
Handling
Anticancer läkemedel. Trastuzumab emtansin är ett antikropp-läkemedelskonjugat som innehåller trastuzumab, en humaniserad monoklonal anti-HER2 IgG1-antikropp, kovalent kopplad till DM1 (maytansinderivat, mikrotubuli-hämmare) via en stabil MCC-tioeterlänk (4- cyklohexan-1-karboxyl) . Emtansine är MCC-DM1-komplexet. Konjugering av DM1 med trastuzumab orsakar selektiv verkan av cytotoxiska läkemedel mot HER2 som överuttrycker tumörceller, vilket ökar den intracellulära koncentrationen av DM1 direkt i tumörcellerna. Efter bindning till HER2 är trastuzumabemtansin receptormedierad internaliserad följt av lysosomal nedbrytning, vilket frigör DM1-innehållande kataboliter (främst lysin-MCC-DM1). Verkningsmekanismen för trastuzumab emtansin förmedlas av aktiviteten av trastuzumab och DM1. Trastuzumab emtansin, liksom trastuzumab, binder till domän IV i den extracellulära domänen (ECD) i receptorn, liksom till Fcy-receptorer och kompletterar C1q. Vidare hämmar den aktiviteten av EC2-domänen i HER2-receptorn, hämmar signalering av fosfatidylinositol-3-kinas (PI3-K) -vägen och förmedlar antikroppsberoende cellulär cytotoxicitet (ADCC) i humana bröstcancerceller som överuttrycker HER2. DM1 binder till tubulin. Genom att hämma tubulinpolymerisation orsakar både DM1 och trastuzumab emtansin celler att stoppas i G2 / M-fasen i cellcykeln, vilket slutligen leder till celldöd genom apoptos. MCC-länken minskar systemisk DM1-frisättning och ökar koncentrationen på målplatsen. Trastuzumab emtansin dekonjugeras och kataboliseras sedan genom proteolys i celllysosomerna. DM1 metaboliseras främst av CYP3A4 och i mindre utsträckning av CYP3A5. T0.5 för trastuzumab är cirka 4 dagar. Trastuzumab emtansin ackumulerades inte efter flera intravenösa infusioner med ett intervall på 3 veckor. Ålder påverkade inte farmakokinetiken för trastuzumab emtansin.
Dosering
Intravenöst. Preparatet ska ordineras av en läkare och administreras under överinseende av vårdpersonal med erfarenhet av behandling av cancerpatienter. Patienter som får trastuzumab-emtansin bör ha HER2-positiv cancer - immunhistokemi (IHC) -poäng 3+ eller förhållande ≥2 vid in situ-hybridisering (ISH). Tester bör utföras med hjälp av in vitro-diagnostiska (IVD) tester med CE-märkning. Om CE IVD-testet inte är tillgängligt bör testet utföras med ett annat validerat test. För att undvika medicinska fel är det viktigt att kontrollera injektionsflaskans etikett för att se till att läkemedlet som bereds och administreras är Kadcyla (trastuzumab emtansin) och inte Herceptin (trastuzumab). Rekommenderad dos är 3,6 mg / kg. ges som en intravenös infusion var tredje vecka (21-dagars cykel). Patienter ska behandlas tills tumörprogression eller uppnått oacceptabel toxicitet. Startdosen ska administreras som en 90-minuters intravenös infusion. Injektionsstället bör övervakas noggrant eftersom läkemedlet kan tränga in subkutant under administrering. Om den tidigare infusionen har tolererats väl kan efterföljande doser ges som en 30-minuters infusion. Patienter ska observeras under infusionen och i minst 30 minuter därefter. Infusionshastigheten bör sakta ner eller avbrytas om patienten utvecklar infusionsrelaterade symtom. Trastuzumab emtansin bör avbrytas vid livshotande infusionsreaktioner. Läkemedel för allergiska / anafylaktiska infusionsreaktioner samt nödutrustning bör finnas tillgängliga för omedelbar användning. Om en planerad dos missas bör den ges så snart som möjligt. vänta inte till nästa cykel. Doseringsschemat bör justeras för att upprätthålla doseringsintervallet på 3 veckor. Nästa dos ska administreras enligt doseringsrekommendationerna. Hantering av symtomatiska biverkningar kan inkludera periodisk avbrytande, dosreduktion eller behandlingsavbrott. Dosreduceringsschema (startdos 3,6 mg / kg): första dosreduktion 3 mg / kg; andra dosreduktion 2,4 mg / kg kroppsvikt; om ytterligare dosreduktion krävs, bör behandlingen avbrytas. Riktlinjer för dosmodifiering för höjningar av ASAT och ALAT: Grad 2 (> 2,5 till ≤5 x ULN) - ingen dosjustering behövs; grad 3 (> 5 till ≤20 x ULN) - använd preparatet om AST / ALT minskar till grad ≤ 2 (> 2,5 till 20 x ULN) - avbryt behandlingen. Dosjusteringsregler för hyperbilirubinemi: grad 2 (> 1,5 till ≤3 x ULN) - använd beredningen när bilirubinnivån har minskat till grad ≤1. (> ULN till 1,5 x ULN), ingen dosjustering nödvändig; Grad 3 (> 3 till ≤10 x ULN) - använd beredningen om bilirubinkoncentrationen sjunker till ≤ grad 1 (> ULN till 1,5 x ULN) och minska sedan dosen (se schema för dosreduktion); Grad 4 (> 10 x ULN) - avbryt behandlingen. Regler för dosmodifiering för trombocytopeni: grad 3 (trombocytkoncentration 25 000 till 3) - använd preparatet när trombocytkoncentrationen är ≤1. (t.ex. ≥75 000 / mm3); ingen dosjustering nödvändig; Grad 4 (trombocytkoncentration 3) - använd preparatet när trombocytkoncentrationen når ≤1. (t.ex. ≥75 000 / mm3) minska sedan dosen (se schema för dosreduktion). Principer för dosmodifiering hos patienter med kammardysfunktion: LVEF 45% - fortsätt behandlingen; LVEF 40% till ≤45% med en samtidig minskning av utkastningsfraktionen med - använd inte preparatet; LVEF-omvärdering inom 3 veckor, avbryt behandlingen om LVEF fortfarande är ≥10 procentenheter från baslinjen; symtomatisk CHF - stoppa behandlingen. Barn och ungdomar: Säkerheten och effekten hos barn och ungdomar under 18 år har inte fastställts eftersom disseminerad bröstcancer (MBC) inte är känd i denna population. Särskilda grupper av patienter. Behandlingen bör avbrytas tillfälligt hos patienter som utvecklar perifer neuropati av grad 3 eller 4 tills den når en grad ≤2; när behandlingen påbörjas kan man överväga att sänka dosen enligt dosreduktionsschemat. Ingen dosjustering krävs för patienter ≥ 65 år; det finns otillräckliga data för att bestämma säkerheten och effekten av behandlingen hos patienter ≥75 år. Ingen dosjustering krävs för patienter med lätt eller måttligt nedsatt njurfunktion. Det potentiella behovet av dosjustering hos patienter med svår njurinsufficiens kan inte fastställas och därför bör dessa patienter övervakas noggrant. Ingen justering av startdosen krävs hos patienter med lätt eller måttligt nedsatt leverfunktion. Trastuzumab emtansin har inte studerats hos patienter med svårt nedsatt leverfunktion. försiktighet bör iakttas vid behandling av patienter med nedsatt leverfunktion på grund av känd levertoxicitet som observerats under behandlingen. Sätt att ge. Preparatet bör lösas upp och spädas av medicinsk personal och administreras som en intravenös infusion. Det får inte ges som en bolus eller snabb injektion.
Indikationer
Monoterapi av vuxna patienter med HER2-positiv, inoperabel lokalt avancerad eller metastaserad bröstcancer, tidigare behandlad med trastuzumab och en taxan, i kombination eller separat. Patienter som tidigare har genomgått behandling för lokalt avancerad eller generaliserad sjukdom, eller som har återkommit under eller inom 6 månader efter avslutad adjuvant behandling.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller andra ingredienser i beredningen.
Försiktighetsåtgärder
Fall av interstitiell lungsjukdom, inklusive lunginflammation, har rapporterats i kliniska prövningar med trastuzumab emtansin; vissa har förknippats med akut andningssvikt eller har varit dödliga. Avbrytande av behandlingen med preparatet rekommenderas hos patienter som diagnostiserats med ILD eller lunginflammation. Patienter med dyspné i vila, förknippade med komplikationer av avancerad cancer och associerade sjukdomar kan ha ökad risk att utveckla lungkomplikationer. Under behandling med preparatet har hepatotoxicitet och allvarliga störningar i levern och gallvägarna, inklusive nodulär regenerativ hypertrofi (NRH) i levern och dödsfall på grund av läkemedelsinducerad leverskada observerats; comorbiditeter och / eller samtidigt läkemedel som är kända för att ha hepatotoxisk potential kan också ha påverkats. Leverfunktionen bör övervakas före behandlingsstart och före efterföljande dosering. Patienter med ALAT-förhöjning vid baslinjen (t.ex. associerad med levermetastaser) är predisponerade för utveckling av leversvikt och har högre risk för levertoxicitet av grad 3-5 eller förhöjda leverlaboratoriska nivåer. Fall av NRH diagnostiserades genom att ta leverbiopsier. Förekomsten av NRH bör övervägas hos alla patienter med kliniska tecken på portalhypertension och / eller cirrosliknande bild som ses vid lever-CT, men med normala serumtransaminasnivåer och inga andra tecken på cirros. Om NRH diagnostiseras ska behandlingen med preparatet avbrytas. Det har inte studerats hos patienter med serumtransaminaser> 2,5 x ULN (övre normalgräns) eller med totalt bilirubin> 1,5 x ULN innan behandlingsstart. Vid serumtransaminasaktivitet> 3 x ULN och samtidigt total bilirubinkoncentration> 2 x ULN bör behandlingen avslutas. Försiktighet bör iakttas vid behandling av patienter med nedsatt leverfunktion. Det finns en ökad risk för vänsterkammardysfunktion under behandlingen. Hos patienter som behandlats med trastuzumab emtansin sågs en minskning av vänster ventrikulär ejektionsfraktion (LVEF) vid 50 år), med ett initialt lågt LVEF-värde (25 kg / m2). Standard hjärtfunktionstester (ekokardiogram eller radiofrekvensangiografi med flera grindar) bör utföras innan behandlingen påbörjas och med regelbundna intervall (t.ex. var tredje månad). Patienternas LVEF vid baslinjen var ≥50% i de flesta kliniska prövningar. Patienter med hjärtsvikt, svåra arytmier som krävde behandling, en hjärtinfarkt i anamnesen eller instabil angina innan 6 månader före randomisering, eller med dyspné i vila på grund av avancerad cancer, uteslöts från studien. I händelse av störningar i vänster ventrikel bör nästa dos skjutas upp eller behandlingen avslutas. Effekten av trastuzumab-emtansinbehandling hos patienter som avbryter behandlingen med trastuzumab på grund av en infusionsreaktion har inte studerats; terapi med detta läkemedel rekommenderas inte i denna patientgrupp. Behandlingen ska avbrytas hos patienter som utvecklar en allvarlig infusionsrelaterad reaktion tills symtomen försvinner. Återstart av behandlingen bör övervägas baserat på klinisk bedömning av reaktionens svårighetsgrad. Behandlingen bör avbrytas vid en livshotande infusionsreaktion. Effekten av trastuzumab-emtansinbehandling hos patienter som avbrutits från trastuzumab på grund av överkänslighet har inte studerats; Användning av trastuzumabemtansin hos dessa patienter rekommenderas inte. Patienter bör övervakas med avseende på överkänslighet / allergiska reaktioner, symtomen kan vara desamma som infusionsrelaterade reaktioner; allvarliga anafylaktiska reaktioner har observerats. Om överkänslighet (med ökade reaktioner vid efterföljande infusioner) utvecklas, bör behandlingen med trastuzumab emtansin avbrytas.På grund av risken för blödningar (inklusive CNS, andnings- och gastrointestinal blödning) bör försiktighet iakttas och ytterligare övervakning övervägas vid samtidig administrering med antikoagulantia eller trombocytläkemedel. Det rekommenderas att antalet blodplättar övervakas före varje dos av trastuzumab emtansin. Patienter med trombocytopeni (≤100 000 / mm3) och patienter som får antikoagulantbehandling (t.ex. warfarin, heparin, lågmolekylära hepariner) bör övervakas noggrant under behandling med trastuzumab emtansin. Trastuzumab emtansin har inte studerats hos patienter med trombocytantal ≤100 000 / mm3 innan behandlingen påbörjades. Vid försämring av trombocytopeni till grad 3 eller högre (3) ska preparatet inte användas förrän toxiciteten minskar till grad 1 (≥75 000 / mm3). I kliniska prövningar med trastuzumab emtansin har perifer neuropati av grad 1, främst sensorisk, inträffat. Patienter med perifer neuropati av grad ≥3 uteslöts från deltagande i kliniska prövningar. Behandling av patienter med grad 3 eller 4 perifer neuropati bör avbrytas regelbundet tills komplikationen minskar till grad ≤2. Patienter bör övervakas regelbundet för tecken på neurotoxicitet. För att förbättra spårbarheten av biologiska preparat bör handelsnamnet för det administrerade läkemedlet vara tydligt skrivet (eller anges) i patientjournalen. Säkerheten och effekten av läkemedlet hos barn och ungdomar under 18 år har inte fastställts eftersom spridning av bröstcancer inte finns i denna population. Det får inte ges som en bolus eller snabb injektion.
Oönskad aktivitet
Mycket vanliga: urinvägsinfektion, trombocytopeni, anemi, hypokalemi, sömnlöshet, perifer neuropati, huvudvärk, blödning, epistaxis, hosta, dyspné, stomatit, diarré, kräkningar, illamående, förstoppning, muntorrhet, buksmärta , utslag, muskuloskeletal smärta, artralgi, myalgi, trötthet, feber, asteni, frossa, ökning av transaminaser. Vanliga: neutropeni, leukopeni, överkänslighet, yrsel, dysgeusi, minnesstörning, torra ögonsyndrom, konjunktivit, synstörningar, ökad lakrimation, vänsterkammardysfunktion, högt blodtryck, dyspepsi, tandköttsblödning, klåda i huden, alopeci, nagelsjukdom , palmar-plantar erytrodysestesi, urtikaria, perifert ödem, ökning av alkaliskt fosfatas, infusionsrelaterade reaktioner (rodnad i huden, frossa, feber, dyspné, lågt blodtryck, väsande andning, bronkospasm, takykardi). Mindre vanliga: lunginflammation (ILD), levertoxicitet, leversvikt, nodulär regenerativ hypertrofi, portalhypertension, extravasation på injektionsstället (erytem, ömhet, hudirritation, smärta eller svullnad). Dessutom har hyperbilirubinemi observerats. I kliniska prövningar inträffade förhöjningar av serumtransaminaser (grad 1-4) under behandling med trastuzumab emtansin och var i allmänhet övergående. Förhöjningen av transaminaser var oftast övergående och toppade vid den åttonde dagen efter dosering. Därefter minskade toxiciteten till grad 1 eller försvann före nästa cykel. Det fanns också en kumulativ effekt (procentandelen patienter med grad 1-2 ALAT / AST-höjning ökade med efterföljande cykler). Hos patienter med förhöjda transaminaser upplevde majoriteten av patienterna grad 1 toxicitetsminskning eller återhämtning inom 30 dagar efter den sista dosen trastuzumab emtansin. Vänsterkammardysfunktion hittades hos 2,2% av patienterna som deltog i kliniska prövningar. I de flesta fall var grad 1 eller 2. LVEF-minskningar asymptomatiska. Grad 3 eller 4. toxicitet observerades hos 0,4% av patienterna, vanligtvis under de initiala behandlingscyklerna (1-2). Allvarliga episoder av blödande händelser (grad ≥3) rapporterades hos 2,2% av alla patienter. Trombocytopeni inträffade hos 24,9% av patienterna i kliniska prövningar och var den vanligaste biverkningen som ledde till att behandlingen avbröts. I kliniska prövningar var incidensen och svårighetsgraden av trombocytopeni högre hos asiatiska patienter. Vissa patienter som utvecklade dessa komplikationer behandlades också med antikoagulantbehandling. Det förekom dödliga blödningsepisoder och allvarliga blödningskomplikationer, inklusive CNS-blödning. 5,3% av patienterna testade positiva för anti-trastuzumab-emtansinantikroppar.
Graviditet och amning
Användning av trastuzumab emtansin till gravida kvinnor rekommenderas inte. Innan de blir gravida ska kvinnor informeras om risken för skada på fostret. Patienter som blir gravida ska dock kontakta läkare omedelbart. Om en gravid kvinna behandlas med trastuzumab emtansin rekommenderas noggrann övervakning av ett tvärvetenskapligt team. Trastuzumab kan orsaka fosterskador eller dödsfall om det ges till en gravid kvinna. Hos barn av gravida kvinnor som behandlats med preparatet har fall av oligohydramnios rapporterats, varav några av dem leder till dödlig lunghypoplasi. DM1, den cytotoxiska komponenten i trastuzumab emtansin, kan vara teratogen och potentiellt embryotoxisk. Kvinnor ska sluta amma innan trastuzumab-emtansinbehandling inleds. Patienter kan börja amma 7 månader efter avslutad behandling. Fertila kvinnor ska använda effektiv preventivmedel under behandlingen med läkemedlet och i 7 månader efter den sista dosen trastuzumab emtansin. Effektiva preventivmetoder bör också användas av män eller deras kvinnliga partner.
Kommentarer
Patienter som får infusionsrelaterade reaktioner ska inte köra bil eller använda maskiner förrän dessa symtom har löst sig. Information utarbetad på grundval av SPC av 13 juli 2017 Den nuvarande produktresumén finns på www.roche.pl.
Interaktioner
In vitro-resultat av metabolismstudier på humana levermikrosomer indikerar att DM1 metaboliseras främst av enzymet CYP3A4 och i mindre utsträckning av CYP3A5. Samtidig användning av starka CYP3A4-hämmare (t.ex. ketokonazol, itrakonazol, klaritromycin, atazanavir, indinavir, nefazodon, nelfinavir, ritonavir, saquinavir, telitromycin och vorikonazol) bör undvikas eftersom detta kan öka DM1-nivåerna och toxiciteten. En alternativ formulering som inte hämmar eller endast hämmar CYP3A4 bör övervägas. Om samtidig användning av starka CYP3A4-hämmare inte kan undvikas, och om möjligt, överväga att fördröja administrering av trastuzumab-emtansin tills CYP3A4-hämmare rensas ur cirkulationen (cirka 3 hämmare-halveringstider). Om en stark CYP3A4-hämmare används samtidigt och behandling med trastuzumab emtansin inte kan försenas, bör patienten följas noggrant med avseende på biverkningar i sådana fall.
Beredningen innehåller ämnet: Trastuzumab emtansin
Ersatt läkemedel: NEJ


















---pseudochoroba-czy-rzeczywiste-zagroenie-wywiad.jpg)